This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison. Information about the course can be found at genetics564.weebly.com/
What is nephrogenic diabetes insipidus(NDI)?
|
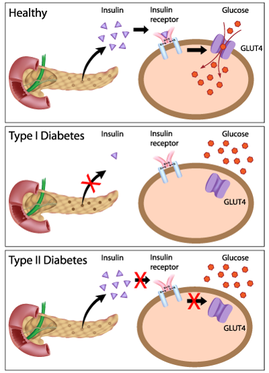
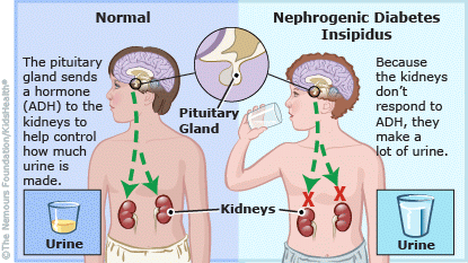
Nephrogenic diabetes insipidus(NDI) is a disease associated with production of large volumes of dilute urine and excessive thirst (1). While diabetes mellitus is caused by disruption of insulin's role within the body, the symptoms of diabetes insipidus are instead caused by a disruption of the role of anti-diuretic hormone(ADH) (2,3). Diabetes insipidus is further subdivided into several disorders based on the reason for ADH disruption. Central diabetes insipidus occurs as a result of an inability of the hypothalamus in the brain to produce sufficient quantities of ADH. ADH promotes the increased absorption of water within the kidney, thus leading to greater urine concentration and lower concentrations of salts in the blood. In contrast, nephrogenic diabetes insipidus is associated with the kidney not responding normally to ADH produced by the hypothalamus(4). Severe dehydration associated with the disease can lead to elevated blood levels of sodium (hypernatremia) and chloride(hyperchloremia) (5).
|
What causes NDI?
NDI can appear as a result of either genetic causes or pre-existing conditions. The most common cases of NDI are due to reactions with medications or due to pre-existing conditions such as sickle cell anemia or protein starvation. NDI can also occur transiently in pregnant women during the duration of their pregnancy(6)
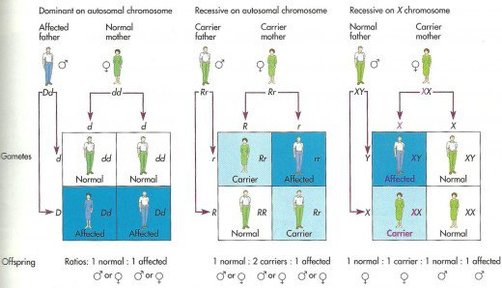
However, NDI can also be inherited in three different manners: X-linked, autosomal dominant, and autosomal recessive. X-linked NDI is associated with mutations in the AVPR2 receptor, which binds ADH (7). X-linked disorders are more common in men then women, since males only have one X-chromosme. This means that if a gene on this X-chromosome is nonfunctional in a male, there is no backup copy. This contrasts with women in that if women have one nonfunctional copy of an X-linked gene than the gene on the other chromosome can usually compensate.
In contrast, the autosomally inherited cases are associated with defects in aquaporin-2(AQP2) (8,9). Autosomally inherited disorders are equally likely to occur in both men and women, since the gene is not on a sex chromosome.
However, NDI can also be inherited in three different manners: X-linked, autosomal dominant, and autosomal recessive. X-linked NDI is associated with mutations in the AVPR2 receptor, which binds ADH (7). X-linked disorders are more common in men then women, since males only have one X-chromosme. This means that if a gene on this X-chromosome is nonfunctional in a male, there is no backup copy. This contrasts with women in that if women have one nonfunctional copy of an X-linked gene than the gene on the other chromosome can usually compensate.
In contrast, the autosomally inherited cases are associated with defects in aquaporin-2(AQP2) (8,9). Autosomally inherited disorders are equally likely to occur in both men and women, since the gene is not on a sex chromosome.
How is NDI treated?
The main treatment for NDI is to have the patient drink large amounts of water to avoid dehydration. Thiazide diuretics can also be administered to increase the concentration of dissolved solids within the urine(10). Thiazides may cause potassium depletion, so they may be used in combination with amilioride to reduce this effect. Urine concentration may also be further increased by prostaglandin inhibitors, as some prostaglandins reduce the effect the ADH has on the kidney.
What is aquaporin-2 (AQP2)?
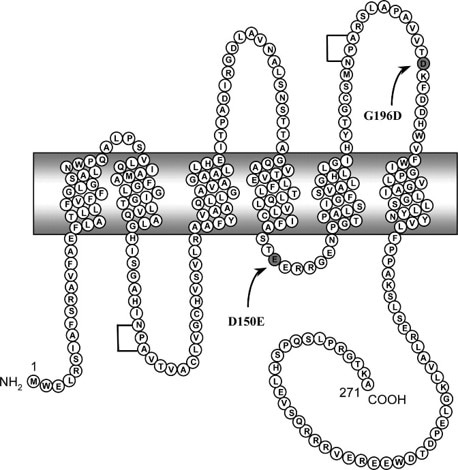
Aquaporin-2 (AQP2) is found within the collecting duct of the kidney, where fluid passes immediately prior to entry into the bladder. ADH signaling in response to lowered blood pressure or higher concentrations of salt in the blood leads to the cells lining the collecting duct to express large amounts of AQP2 (11). This in turn promotes water absorption back into the body to increase blood volume and increase urine concentration.
References
1. Nephrogenic diabetes insipdus foundation (n.d.). Description. Retrieved from www.ndif.org/pages/4-Description . Accessed May 12, 2017.
2.Introduction to diabetes mellitus. Frits Holleman. https://www.diapedia.org/introduction-to-diabetes-mellitus/1104085113. Accessed May 11th,2017.
3. Diabetes insipidus-central. 2015. https://medlineplus.gov/ency/article/000460.htm. Accessed May 12,2017.
4.Nephrogenic Diabetes Insipidus. National Organization for Rare Disorders. 2016. https://rarediseases.org/rare-diseases/nephrogenic-diabetes-insipidus/. Accessed May 11th, 2017
5. Diabetes Insipidus. The Pituitary Foundation 2017. https://www.pituitary.org.uk/information/pituitary-conditions/diabetes-insipidus/. Accessed May 11th,2017
6. Ejmocka-Ambroziak, A., Grzechocinska, B., Jastrzebska, H., Kochman, M., Cyganek, A., Wielgos, M., & Zgliczynski, W. (2015). Gestational diabetes insipidus. Neuroendocrinology Letters, 36(5), 410-413.
7.Seibold, A., Rosenthal, W., Bichet, D. G., & Birnbaumer, M. (1993). The vasopressin type-2 receptor gene - chromosomal localization and its role in nephrogenic diabetes-insipidus. Regulatory Peptides, 45(1-2), 67-71.
8. Vanlieburg, A. F., Verdijk, M. A. J., Knoers, V., Vanessen, A. J., Proesmans, W., Mallmann, R., . . . Deen, P. M. T. (1994). Patients with autosomal nephrogenic diabetes-insipidus homozygous for mutations in the aquaporin-2 water-channel gene. American Journal of Human Genetics, 55(4), 648-652.
9. Mulders, S. M., Bichet, D. G., Rijss, J. P. L., Kamsteeg, E. J., Arthus, M. F., Lonergan, M., . . . Deen, P. M. T. (1998). An aquaporin-2 water channel mutant which causes autosomal dominant nephrogenic diabetes insipidus is retained in the Golgi complex. Journal of Clinical Investigation, 102(1), 57-66. doi:10.1172/jci2605
10. Mizuno, H., Fujimoto, S., Sugiyama, Y., Kobayashi, M., Ohro, Y., Uchida, S., . . . Togari, H. (2003). Successful treatment of partial nephrogenic diabetes insipidus with thiazide and desmopressin. Hormone Research, 59(6), 297-300. doi:10.1159/000070629
11.Deen, P. M. T., Verdijk, M. A. J., Knoers, N., Wieringa, B., Monnens, L. A. H., Vanos, C. H., & Vanoost, B. A. (1994). Requirement of human renal water channel aquaporin-2 for vasopressin-dependent concentration of urine. Science, 264(5155), 92-95.
Zach Dumar
[email protected]
Page last updated:May 12th 2017
Additional student pages can be found at genetics564.weebly.com
[email protected]
Page last updated:May 12th 2017
Additional student pages can be found at genetics564.weebly.com

|

|

